“Why, we can hardly call it a complaint, Miss Ruthyn. I look upon it he has been poisoned-he has had, you understand me, an overdose of opium; you know he takes opium habitually; he takes it in laudanum, he takes it in water, and most dangerous of all, he takes it solid, in lozenges. I’ve known people take it moderately. I’ve known people take it to excess, but they were all particular as to measure, and that is exactly the point I’ve tried to impress upon him. The habit, of course, you understand is formed, there’s no uprooting that; but he won’t measure-he goes by eye and sensation, which I need not tell you Miss Ruthyn, is going by chance; and opium, as no doubt you are aware, is strictly a poison; a poison no doubt, which habit will enable you to partake of, I may say, in considerable quantities, without fatal consequences, but still a poison; and to exhibit a poison so, is, I need scarcely tell you, to trifle with death.”
Uncle Silas -Joseph Sheridan Le Fanu
For many years now there has been nothing but bad news about opioids. In 2021 over 100,000 people in the US died from drug overdoses. It’s a huge problem. But we tend to ignore something that is equally important about opioids: they are the most powerful substances humanity has ever encountered for the treatment of pain. Given the fact that pain is the most basic of all human complaints, one could certainly make the case that opium and its derivatives are the most important drugs in all of medicine. Consider the fact that the entire practice of anesthesiology depends on the use of opioids for performing surgery. No opioids would mean no heart operations, no joint replacements, and an enormous number of other procedures would simply be impossible. Opioids are really an extreme example of the dichotomous nature of all drugs, the fact that they can produce both beneficial and toxic effects. We know that opioids are extremely powerful analgesic drugs and are widely used in medicine for this and other purposes. On the other hand, take a little too much and they will stop you breathing (respiratory depression), and they are certainly extremely addictive. As we shall see, similar things are true for many other important drugs which humans originally discovered through their exploration of the natural world.
The fact that all drugs can produce both beneficial and toxic effects was clearly recognized by doctors and pharmacologists dating back to antiquity. In the sixteenth century this idea was encapsulated by the alchemist Paracelsus who said, “All things are poison, and nothing is without poison; the dosage alone makes it so a thing is not a poison.” And it’s certainly the case that we can often adjust the dose of a drug so that its beneficial effects are apparent and its toxic side effects are reduced. However, it isn’t always possible to do this very easily. The use of opioids, for example, is extremely complicated because similar doses can produce both analgesia and problems such as addiction.
Separating the desirable and undesirable effects of a drug is one of the most important challenges in medicine and is a major concern of the science of pharmacology. There are generally three approaches to doing this. Taking Paracelsus’ advice and changing the dose may help. Changing the route of drug administration is another possibility. Finally, and most profoundly, we can attempt to use the tools of organic chemistry to alter the molecular structure of the drug hoping to accentuate its therapeutic actions and reduce its toxic effects. Opioid drugs are a fantastic example of this challenge. Since the dawn of modern science in the seventeenth century there have been constant attempts to tame opium but, as will be clear from the current epidemic, it’s something we have failed to do. But that doesn’t mean we haven’t tried. Indeed, scientists have devoted a huge amount of time and effort to solving this problem over hundreds of years.
The dawn of modern chemistry
In the seventeenth century, during the English Civil War, the city of Oxford had become a mecca for many of the individuals who were to spearhead the coming Scientific Revolution, allowing science to free itself from the antiquated ideas that had shackled its progress since the fall of the western Roman Empire in the fifth century CE. These men organized themselves into clubs for the purpose of conducting experiments and discussing their results. One club, which met on the premises of an apothecary, was particularly interested in applying new ideas from the emerging science of chemistry to medicine. One of the most significant members of this group was Sir Robert Boyle, known to many today as “The Father of Chemistry”. Boyle was a wealthy aristocrat, the 14th child of the 1st Earl of Cork. He became interested in chemistry as a young man and had the financial wherewithal to carry out his own independent studies.
Boyle was one of the earliest members of the Royal Society founded in 1660, the first organization of its type devoted to fostering the progress of experimental science. Through the efforts of Boyle and several of his colleagues, the experimental method became the most important way of establishing scientific truths and experiments were increasingly used for understanding the elemental nature of matter and how things could be transformed from one substance into another.
In the mid-1650s, Boyle completed an article entitled ‘An Essay of Turning Poisons into Medicines’. In his essay Boyle commented on the potential therapeutic benefits of a number of otherwise toxic agents including mercury, antimony, arsenic, viper venom and opium*.
(* Mercury and arsenic, for example, were used to treat syphilis, although they are clearly very toxic substances. Balancing their diverse properties was not easy and often the cure was as bad or worse than the disease. Eventually the toxic effects of arsenic would be reduced by using organic chemistry to modify its properties).
Boyle was very devout and thought that revelations about the beneficial effects of chemicals were the result of divine providence. He was determined to use newly acquired knowledge about chemistry in the service of the Almighty. Boyle became interested in the possibility that the diverse effects of a drug might depend on the way it was administered. People in England at the time were aware of the effects of arrow poisons and snake venoms used by American Indians, and many were fascinated by their properties. Boyle showed that the poisonous effects of a viper were only apparent when it bit you so that its poison entered your bloodstream. Just eating a viper’s venom was not poisonous. He demonstrated this as follows –“I have been induced to suspect upon this Experiment; That dissecting some live Vipers, there came in accidentally a strange Dog, to whom I gave the Head, Tail, and Gall (which are the parts supposed to contain the Poyson) of one of them, and the Head and Gall of another, wrapt up in meat; after which, I locked the little Dog up in my own Chamber, and watched him, but found not that he was sick, or offerd to vomit at all, but onely lap’d up greedily some drink which he espyd in the Room; nor was he alone very jocund, for divers hours that I kept him in, but liked his entertainment so well, that he would afterwards, when he met me in the Street, leave those that kept him to fawn on and follow me.” Boyle was in two minds about doing experiments on animals, but it seems that there was a constant stream of dogs that wandered into his laboratory, and he was certainly prepared to use them.
Directing medicines specifically to the bloodstream rather than taking them by mouth was a novel idea that was in keeping with the cutting-edge science of the time. While Boyle was living in Oxford, we know he met the great William Harvey who had also taken up residence there and that he was aware of Harvey’s famous research describing the circulation of the blood, one of the great achievements in the history of science. So, it seems that the idea of an intravenous injection was very much on his mind as a possible route for effective drug administration as, according to Harvey’s ideas, the circulating blood would be an efficient way of distributing the drug around the body.
Boyle went on to consider the effects of opium which was well known to have poisonous properties as well as therapeutic effects. Opium was certainly a wonderful drug for treating pain, cough, diarrhea, and other ailments. However, it could also make you vomit, and could easily kill you by depressing your breathing. What would be the effect of administering opium intravenously? Would it still work when administered by this route? Perhaps its effects would be more favorable when given in this manner so that it would retain its pain killing properties but not its detrimental effects on breathing? It had been shown by Paracelsus in the sixteenth century that the active constituents of opium could be extracted into alcohol to make a tincture known as laudanum and perhaps this might be injected directly into the blood? In order to carry out this experiment, in 1656 Boyle collaborated with the young Christopher Wren, later the architect of St Paul’s cathedral in London. First, they dissolved some opium in warm sack (sherry) and then injected it intravenously, using the quill of a pen, into yet another dog that had wandered into Boyle’s laboratory. Boyle observed that –“getting into the mass of Blood . . . was quickly, by the circular motion of That, carry’d to the Brain, and other parts of the Body. So that we had scarce unty’d the Dog . . . before the Opium began to disclose its Narcotick Quality, and almost as soon as he was upon his feet, he began to nod with his head, and faulter and reel in his pace, and presently after appear’d so stupifi’d, that there were Wagers offer’d his Life could not be sav’d”. So, it seemed that the effects of intravenous laudanum were similar to those observed when drinking it. As it turned out the dog did recover. Recovery was achieved by causing the dog -‘to be whipp’d up and down the Neighboring Garden, whereby being kept awake, and in motion, after some time he began to come to himself again’. The dog was ‘carefully tended, began to grow fat so manifestly that 'twas admired: But I could not long observe how it far'd with him. For this Experiment, and some other tryals I made upon him, having made him famous, he was soon after stolen away from me.” An Internet star avant la lettre!
By performing experiments of this type, Boyle and his canine assistants provided key experimental data about the possibility of effectively delivering drugs by the intravenous route. Soon they were pushing their method further and carrying out the first blood transfusions. A new therapeutic paradigm had been born.
Although Boyle had attempted to separate the different properties of opium based on the way it was administered to a live animal, he had not considered the alternative possibility that this same separation of properties might result from actual chemical transformation of the drug itself. Such a possibility would require a better understanding of the precise chemical constituents of opium which was not available at the time. Boyle was one of the leading scientists who believed in the corpuscular nature of the elements as the underlying building blocks of matter, but in the late seventeenth century there was no real understanding of the true nature of the elements or the fundamental laws of chemistry that governed the ability of one substance to react with another.
The rise of atomic theory
The identity and chemically unique properties of the elements began to reveal themselves in the period following Boyle. Eventually, new experimental results and ideas led to Dalton’s atomic theory which appeared in its mature form in the first decade of the nineteenth century. It held that all elements are made up of small indivisible particles called atoms. The atoms of a given element are all the same with respect to mass and other properties but differ from the atoms of every other element. Chemical combination occurs when the atoms of two or more elements form a stable union. The theory was further developed, particularly by Berzelius, who introduced the modern symbolic terminology for describing the elemental structure of matter leading to the language of chemistry that became uniformly accepted and helped conceptualize chemical reactions and the structures of different molecules. Nevertheless, some areas of chemistry advanced more quickly than others and it was the chemistry of living materials or “vegetable chemistry”, as it was known, that initially lagged behind. For a chemical understanding of living matter to advance, scientists would have to come to grips with the properties of what is perhaps the most remarkable element of all, and that is carbon.
The existence of carbon has been known since ancient times in the form of things like soot and charcoal. During the rapid advances in chemistry during the seventeenth and eighteenth centuries came the realization that, like many other substances, carbon is an element. However, the chemical reactivity of carbon is particularly wide ranging; it can form four separate bonds with other atoms including other carbon atoms, hydrogen, oxygen, and nitrogen, giving it an extremely wide choice of chemical partners. Hence, carbon can form molecules that range from being small in size such as methane or carbon dioxide, to extremely complex polymers consisting of thousands of atoms. It is this unique ability to construct gigantic molecular structures under energetic conditions that prevail on Earth, that allows carbon to form the basic building blocks of living matter such as proteins, nucleic acids, sugars, and fats, as well as the metabolic intermediates that enable the production of the biological energy that fuels life. Indeed, in many respects it is carbon’s unique reactivity that enables the existence of life in the form that we know it. Hence, if we want to chemically operate on living materials so as to transform them, we need to understand the chemistry of carbon. The results of such knowledge have had profound consequences for the understanding of the fabric of Nature in general. Indeed, the entire range of carbon-based chemistry, usually referred to as “organic chemistry”, including disciplines like biochemistry as well as the fossil fuel industry, is an enterprise which is at least as large as all other types of chemistry put together.
As chemistry began to advance rapidly at the end of the eighteenth century there were relatively few carbon-based substances that had been obtained in pure form. Alcohol was one of these and acetic acid from vinegar was another, both obtained through the process of distillation which had been extensively developed in association with medieval alchemical practices. However, towards the end of the 18th century Carl Wihelm Scheele, one of the discoverers of oxygen, also discovered a wide range of organic acids including oxalic, malic, tartaric, citric, lactic, uric and gallic acids. Glacial acetic acid, meaning it was free of water, was also purified at the same time. All these carbon containing reagents could now be used as building blocks for performing chemical reactions in the organic sphere.
Scheele’s discoveries were organic or “vegetable acids”. Organic nitrogen containing “vegetable bases” or alkalis would soon follow. In 1805 a German pharmacist called Friedrich Wilhelm Serturner, isolated a pure white crystalline material from opium. He tested it on several students and dogs and found that it readily put them to sleep; so, he called it morphium after Morpheus the God of dreaming from Ovid’s Metamorphosis. The name was subsequently changed to morphine. Soon a long list of similar nitrogen containing molecules were isolated from other natural sources. These “natural products” included another substance isolated from opium named codeine, which produced similar effects to morphine. Caffeine, strychnine, quinine, nicotine, and many other substances were also isolated from a variety of plants in the early nineteenth century. Because, like morphine and codeine, all these substances had a basic, alkaline character, the entire class of molecules became known as alkaloids.
The biological properties of crude opium as used throughout history by investigators like Robert Boyle and Paracelsus can be explained by its content of the pure molecules morphine and codeine. As far as morphine itself was concerned, it proved to be a large and complex organic molecule although its exact chemical structure, that is to say the shape that its constituent atoms form in 3-dimensional space, wasn’t determined until 1925. Nevertheless, following Serturner’s work, together with the increasing availability of a number of organic chemical reagents, morphine was ripe for attempts to use chemical reactions to alter its structure and, hopefully, its properties.
Dyes and pharmaceuticals
In coming years, the major enterprise that would be interested in the modification of morphine would be the pharmaceutical industry. But first there had to be a pharmaceutical industry, and this wasn’t the case in 1805 when pure morphine was first isolated. The advent of the pharmaceutical industry proceeded in a somewhat unexpected manner. One abundant source of new organic molecules in the early nineteenth century was the coal mining industry, particularly coal tar which resulted from the coking of coal. In 1856 a young English chemistry student named William Perkin, he was only 18, was given the task of making synthetic quinine as part of his homework. This proved to be beyond his (or anybody else’s) capabilities at that time. Nevertheless, he gave it a try in his home laboratory and observed that by treating the organic molecule aniline, originally obtained from coal tar, with potassium dichromate, he could produce a beautiful pink/purple substance which could act as a dye. It was the first synthetic dyestuff ever created and he named it mauve. Prior to the discovery of mauve, all dyes had to be isolated from natural sources, a very labor-intensive process. The synthesis of many new aniline-based dyes quickly followed. Soon after Perkin’s discovery, a highly profitable synthetic dye making industry was established which helped to energize the burgeoning science of organic chemistry. At some point the dye making companies thought about diversifying their numerous new chemical discoveries and observed that some of their products had useful pharmacological activities, particularly for lowering fevers. Soon many of the dye making companies had also become pharmaceutical companies. This was particularly true in Germany where companies like Bayer, Hoechst, Merck (originally an apothecary) and BASF (Badische Analin und Sodafabrik) were established. The future of morphine would be very much in their hands.
At the dawn of the 19th century which saw the rise of the Romantic movement, recreational drug taking became a much more widespread activity and it started to become generally appreciated that morphine’s addictive properties were really an important medical issue. Like Boyle before them, pharmaceutical chemists began to think of ways of getting round the problem of trying to separate the adverse from the useful effects of morphine. Now, they had new methods available to them for potentially achieving this goal; the use of the tools of organic chemistry for transforming the molecular nature of the drug. Surely, changes to the structure of morphine would yield new molecules that possessed desirable properties such as killing pain but were free of addictive potential, respiratory depression, and other issues?
The first attempts to chemically transform morphine and to remove its poisonous properties were very simple and were based on its treatment with one of the strong mineral acids such as sulfuric or hydrochloric acid. The results of this experiment were first reported by the Finnish chemist Adolf Edvard Arppe in 1845 and were certainly unexpected. This simple chemical reaction not only changed the structure of the morphine molecule but also completely transformed its biological activity from being an opioid drug to being a possible cure for opiate addiction, an extremely dramatic example of what can be achieved by applying the rules of chemistry to the products of Nature.
The molecular product that was obtained by Arppe was initially named sulfomorphide. It didn’t attract a lot of interest until it was also synthesized by the English chemists Augustus Matthiessen and Charles Alder Wright who obtained the same product in 1869.They named the resulting molecule apomorphia or, subsequently, apomorphine; the “apo” designation indicating that it was from morphine rather than like it .Indeed ,its properties turned out to be completely different-one investigator commenting ‘‘in spite of its name, apomorphine is no more like morphine than sawdust is like sugar’’. True enough. Apomorphine possessed none of the euphoric psychological or analgesic effects associated with morphine. Its actions were entirely unpleasant. Prominent among these was its powerful emetic effect, something that it did share with morphine. Hence, the synthesis of apomorphine had indeed deconstructed the effects of morphine. A new substance had been created where the analgesic and other positive effects were lost leaving the unpleasant emetic effects; the exact opposite of the type of result that had been hoped for. Indeed, the overall effects of taking apomorphine were described as ‘‘Lassitude, weakness, frequent headaches, constant nausea, and occasional sudden attacks of vomiting’. Nevertheless, the experiment wasn’t a failure. As we shall see, apomorphine was to have a most interesting future.The reason for the differences in the properties of morphine and apomorphine can be understood by examining the latter’s chemical structure. Heating morphine with a strong acid promotes its dehydration and chemical rearrangement. In other words, apomorphine is a dehydrated form of morphine. In 1869, apomorphine’s chemical formula (C17H17NO2) was found to be the same as morphine’s formula (C17H19NO3) minus a water molecule (H2O), but its exact structure was only elucidated in 1902. This was very revealing and can be easily appreciated even by a non-chemist (Figure 1). The two hydroxyl groups (OH) present in the morphine molecule, situated on rings A and C, are key structural features that determine its opioid like activity. Blocking one of these, as in codeine, reduces the activity of the molecule and, as we will discuss, blocking both of them destroys it completely. If one looks at the structure of apomorphine, it seems similar to morphine at first glance. There are some differences such as the fact that it only has 4 rings instead of 5, but it still has two hydroxyl groups. However, the position of these two groups has now changed. Instead of having one on the A ring and one on the C ring, they are both on the A ring. This arrangement of hydroxyl groups is known as catechol and, as it turns out it, gets you into a completely different realm of pharmacology and physiology. In 1965, Anton Marie Ernst was the first to draw attention to the structural relationship between apomorphine and dopamine an important neuronal messenger (“neurotransmitter”).
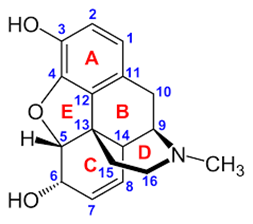

One can see the outline of the dopamine structure snaking its way through the structure of apomorphine (Figure 2) and, it is the activity of apomorphine at dopamine receptors, rather than at opioid receptors, that is responsible for its spectrum of biological effects. A simple chemical reaction had acted as a biological switch completely changing the pharmacology of morphine into something else that was still powerful but completely different.

Indeed, it was the entirely unpleasant effects of apomorphine that gave rise to its initial utility. Originally used as an emetic (i.e., to induce vomiting) the possible use of apomorphine as a modifier of behavior was first explored by pharmacologist Erich Harnack, an ethnic German born in Estonia, whose groundbreaking research in the late nineteenth century led to apomorphine’s first association with the treatment of alcohol dependence. Harnack used the aversive effects of apomorphine by administering the drug together with alcohol to produce a negative Pavlovian type of behavioral conditioning aimed at stopping alcoholics drinking. The treatment was found to be very effective and became the basis of the Keeley Cure for alcoholism that was popular initially in the US and then throughout the world in the early 20th century.
The apomorphine cure
One physician who became extremely interested in using this treatment on his patients was John Yerbury Dent. Dent was a young doctor working in North London’s St Pancras hospital particularly with the local population of alcoholics. At that time the idea of treating alcoholism was entirely novel. The usual treatment for alcoholics was to pump their stomachs and then release them back on to the streets where they would eventually repeat their behavior or die. But Dent was a creative thinker: he liked to think out of the box and was attracted by the controversial idea that alcoholism was a disease and, if it was a disease, it could be treated as such using a well thought out medical and pharmacological approach as typified by Harnack’s studies.
Dent began experimenting with apomorphine and found that when he gave it to his alcoholic patients it really did seem to help. Although he initially agreed with the idea that Pavlovian conditioning was responsible for the drug’s beneficial effects, he began to change his mind. It seemed to him that apomorphine had a more complicated effect than just producing an aversion to drinking. He proposed that apomorphine specifically reduced the state of craving for alcohol and helped the patient through the worst phases of alcohol withdrawal. ‘‘This is one of the very few treatments in which the patient can expect a miracle’’ Dent proclaimed. As time went on Dent and others started to use apomorphine to treat other substance abuse disorders including addiction to opioids, barbiturates, and tobacco. It was even used to treat “homosexuality”, considered a psychiatric disorder at the time, by attempting to induce nausea together with electric shocks while presenting the patient with pictures of male nudes. Dent became interested in the wide-ranging possibilities for using apomorphine, at least as far as substance abuse was concerned. Of course, there were others who disagreed with Dent’s methods. But some people were sold on them. One of them was William Burroughs.
William Seward Burroughs was born in St Louis Missouri in 1914. He was the scion of a wealthy family, being the grandson of the inventor of the Burroughs calculating machine and founder of the Burroughs corporation. Together with Jack Kerouac and Allen Ginsburg, Burroughs was to become one of the seminal influences on the Beat generation of the 1950s. His life, as reflected in his books, was one of a cultural agent provocateur constantly pushing the boundaries of the acceptable. He represented a triple threat of drug addiction, homosexuality and extreme literary experimentation which certainly never endeared him to middle class tastes. Burroughs once declared that the goal of his novels was to “extinguish all rational thought”. Enough said. After a somewhat disjointed college experience, Burroughs began his peripatetic life by moving to Mexico, where he accidently killed his wife while attempting to shoot an apple off her head William Tell style during a drunken binge, and then to Paris and Tangier. Here in the late 1950s, he was on the verge of literary celebrity looking for a publisher of his best-known work Naked Lunch. He had become addicted to heroin and other opioids such as Eukodol (oxycodone) for many years and, it appears, wanted to do something about it. But so far, all treatments had failed. Then Burroughs heard about John Dent and moved to London so that he could undergo apomorphine therapy. And apparently it worked. In Burroughs own words-
“I was living in Tangier in 1957, and I had spent a month in a tiny room in the Casbah staring at the toe of my foot. The room had filled up with empty Eukodol* cartons; I suddenly realized I was not doing anything. I was dying. I was just apt to be finished. So, I flew to London and turned myself over to Dr. John Yerbury Dent for treatment. I’d heard of his success with the apomorphine treatment. Apomorphine is simply morphine boiled in hydrochloric acid; it’s nonaddictive. What the apomorphine did was to regulate my metabolism. It’s a metabolic regulator. It cured me physiologically. I’d already taken the cure once at Lexington, and although I was off drugs when I got out, there was a physiological residue. Apomorphine eliminated that. I’ve been trying to get people in this country interested in it, but without much luck.”
(*oxycodone -see below)
And -
“The apomorphine cure is qualitatively different from all other methods of cure. I have tried them all […] I can say definitively that I was never metabolically cured until I took the apomorphine cure. […] I suggest that research with variations of apomorphine and synthesis of it will open a new medical frontier extending far beyond the problem of addiction.” (Figure 3).

Burroughs reported his experiences at length in various essays ranging from one rather formal article in the British Journal of Addiction to another designated Health Bulletin APO-33, a cut-up text published by the FuckYou press (Figure 4). He certainly attributed a major turnaround in his health and ability to write to his apomorphine therapy commenting- “At the time I took the apomorphine cure I had no claims to call myself a writer and my creativity was limited to filling a hypodermic. The entire body of work on which my current reputation is based was produced after the apomorphine treatment and would never have been produced if I had not taken the cure and stayed off junk.”

It seems that Burroughs was never completely cured of his opioid addiction, arguably nobody ever is. He needed further treatment at intervals. But something had clicked in his brain. The question of course is what? According to Burroughs and others at the time apomorphine acted as a “hindbrain metabolic regulator”-whatever that means. But there is certainly a scientific rationale according to which one might postulate a mechanism. First one might consider the question- “why are opioids and other drugs addictive”? The answer appears to be due to their actions on a specific nerve pathway that runs from a midbrain nucleus named the ventral tegmental area (VTA) and innervates brain structures in the forebrain such as cortex and limbic system. This pathway has an important role in evaluating the rewarding aspects of different stimuli and is sometime called the brain’s “reward circuit”. Activation of this pathway by desirable stimuli like eating and sex produces the rewarding effects of such activities. Drugs like morphine, cocaine, nicotine, and other addictive substances hijack and hyperactivate these brain circuits in what is essentially an irreversible manner producing incessant craving. One interesting thing to note about these reward neurons is that they use dopamine as a neurotransmitter. It seems reasonable to suppose, if opioids can activate these neurons, something that can inhibit this activation could have a beneficial effect on drug induced craving. And, as a drug that works on the dopaminergic system, apomorphine is in a position to produce precisely such effects. There are several mechanisms by which this could happen. This could well be the reason for its beneficial effects when treating opiate addiction.
In spite of Burroughs’ extensive proselytizing, apomorphine therapy never really caught on. Burroughs, who tended to be rather paranoid, assumed that it wasn’t in the financial interests of pharmaceutical companies to find things that would cure addiction. As a therapy apomorphine was controversial and not everybody found it to be successful. Moreover, other therapeutic paradigms were introduced for treating addiction. These generally fell into the category of opioid maintenance or replacement therapy in which another opioid, typically methadone, buprenorphine or levacetylmethadol (LAAM) is substituted for heroin and the patient is gradually weaned off the drug. However, as we have seen, apomorphine’s mechanism of action is quite different from this and it may be that it produces a more curative effect. Even today apomorphine treatment has its advocates but much more work still needs to be done to examine its true effectiveness and utility. Nevertheless, it does seem like an interesting lead.
This then was the beginning of the story of humanity’s very first attempts to alter Nature through chemistry. A simple chemical reaction completely altered the properties of morphine creating a new drug that might act as its own antidote. But that was nowhere near the end of the story.
The discovery of heroin and oxycodone
Following the synthesis of apomorphine, pharmacologists have produced literally thousands of new opioid drugs over the years that have similar effects to morphine and may be more useful in one respect or another, but none of them are more effective as pain killers and, unfortunately, none of them are free of the addictive and other side effects of morphine. Nevertheless, just like Sir Robert Boyle in the seventeenth century and the production of apomorphine in the nineteenth century, there have been many subsequent attempts to free morphine of its addictive liability. And why not? Such things were achieved around the same time with other addictive drugs. Take cocaine for example. Cocaine is a powerful and highly addictive psychostimulant drug and is also an extremely effective local anesthetic, which is why your mouth goes numb when you chew coca leaves. Can chemistry be used to separate these two characteristics? Chemically speaking cocaine is what is known as a tropane alkaloid (Figure 5).

One half of the molecule has a tropane structure, and the other half has a benzoic acid structure. What happens if you cut the molecule in half using chemistry? It turns out that neither half by itself is capable of producing the addictive psychostimulant effects observed with cocaine; but the benzoic acid portion, particularly with a little extra chemical coaxing, can still produce excellent local anesthetic effects. Hence cocaine begat procaine (Novacaine), lidocaine, and a whole list of other “caine” derivatives. Surely, then, this should be possible with morphine? Play around with the molecule using chemistry and you would separate analgesia from addiction, euphoria from emesis and stopping coughing from stopping breathing. Unfortunately, even though this strategy had worked with cocaine, it has never really worked with morphine. Following the production of apomorphine there have been innumerable of attempts to “improve” morphine but none of them have ever been successful. Thousands of powerful opioid analgesics have been produced but none of them have lost their poisonous qualities or addictive potential. Nevertheless, there have been some very interesting drugs produced which have had important medical and cultural consequences.
Although the synthesis of apomorphine was the first example of the chemical transformation of morphine, several other interesting experiments followed shortly thereafter. Of particular interest, was a report by the English chemist Charles Alder Wright in 1874 on the treatment of morphine with acetic anhydride (derived from acetic acid), a powerful organic reagent for adding acetyl groups to other susceptible organic molecules. He succeeded in producing both mono and diacetyl morphine which he demonstrated had properties that were similar to those of morphine and codeine. His report made little impact. But, as we shall see, that would change.
Nineteenth century efforts to achieve Boyle’s aim of turning “poisons into medicines” were not only directed at morphine. Another substance of potential interest was salicylic acid which could be obtained from willow bark. Willow bark preparations had an ancient history of being able to lower fevers, but they were very hard on the stomach and difficult to tolerate. Perhaps chemical modification might help with this by separating the intolerable stomach-churning effects from the beneficial fever reducing properties? In 1853 a French scientist named Charles Gerhardt attempted to modify salicylic acid by treating it with acetyl chloride. The experiment was a bit of a mess, but he was able to make some acetylsalicylic acid although he wasn’t able to examine its pharmacological properties. Nevertheless, he had succeeded in doing something very interesting. Some three and a half decades later a similar idea occurred to scientists at the Bayer pharmaceutical company in Germany. The company already had a major interest in fever reducing drugs and one of their chemists named Felix Hoffman had read Gerhardt’s publication and decided to try the same thing, but this time using acetic anhydride which is a much more efficient way of transferring organic acetyl groups to susceptible substrates. This resulted in a more efficient synthesis of acetylsalicylic acid (ASA). ASA proved to have powerful fever reducing properties and it was much easier on the stomach than anything Bayer had previously produced. As far as a name was concerned, the company chose to use Spirea, the genus of plants including willow from which salicylic acid, otherwise known as spiric acid, was derived. Adding an “a” to the beginning was used to indicate the result of acetylation resulted in “aspirin”-a drug for the ages.
But Hoffman and his colleagues weren’t finished yet. Perhaps the beneficial effects of acetylation might also apply to the treatment of morphine? Hence two weeks after making aspirin the Bayer scientists tried the same chemical reaction but substituting morphine for salicylic acid and rediscovered what Alder Wright had previously demonstrated, the synthesis of diacetylmorphine. Testing this new product on various animals and themselves, the scientists recognized that they had made a powerful new opioid drug that made them feel strong or “heroisch” in German and so decided to call the new drug heroin- ending what was surely one of the most fascinating fortnights in the history of the pharmaceutical industry. In 1898 Bayer put their two new acetylation products aspirin and heroin on the market. There are plenty of advertisements promoting them together; one a fever reducer and the other a “non-addictive” alternative to morphine for use as a cough suppressant. Well, at least they were right about one of them.

As a matter of fact, heroin is a completely inactive opioid drug. Both of its important hydroxyl groups are blocked. But heroin is very soluble in the body and enters the brain much more easily than morphine. Once there, the two acetyl groups are removed regenerating morphine which is now responsible for the observed effects. Heroin is a “prodrug”, a ballistic missile system for the rapid and efficient delivery of morphine into the brain (Figure 6). Following the introduction of heroin, addicts started to appear in New York and other cities in the opening decades of the twentieth century. These addicts often sold old pieces of junk to get money for buying drugs and became known as “junkies”-a worldwide drug addiction problem was taking root.
Bayer’s attempts to improve morphine’s properties proved to be one of the opening shots in what was to be the protracted chemical war against the evils of morphine. Realizing the increasingly widespread nature of the opioid addiction problem, in 1929 the US government funded one of the first systematic studies of morphine aimed at producing new chemical structures that would dissociate analgesic effectiveness from side-effect and addiction liability. For the next three decades there were a large number of additional attempts to chemically modify the structures of morphine and codeine making small changes to the naturally occurring parent molecules. These new drugs are usually described as “semi-synthetic” derivatives of morphine. The major skeleton of the molecule is still based on the structure of morphine or codeine, but a small tweak has been added to them using organic chemistry. The drugs produced included dihydrocodeine, differing from codeine only in saturation of one double bond, hydrocodone, having, in addition, one hydroxyl replaced by oxygen and hydromorphone the equivalent derivative of morphine (Figure 7). Many of these semi-synthetic opioids turned out to be effective drugs but were very similar to morphine and codeine in both their beneficial and side effect profiles. On the other hand, many of them would find useful roles in medicine and the future of oxycodone, in particular, would be very interesting indeed.

Oxycodone was first synthesized in Germany in 1916 by Freund and Speyer. They used a trick which turned out to be extremely useful when exploring the chemistry of morphine, which was to use thebaine for their starting material (Figure 8). Thebaine, which is also found to occur naturally in crude opium, has a structure that is very close to that of morphine, but has no opioid like properties because both of its important hydroxyl groups are blocked. However, sometimes thebaine is easier to use in a chemical synthesis than morphine itself and has been very useful for this purpose throughout the years.

The opioid epidemic
By 1917 oxycodone was being widely used as a substitute for morphine for clinical purposes under the name Eukodal. During the Second World War we know that Adolf Hitler was frequently dosed with it together with a cocktail of vitamins stimulants, laxatives, and various other things to “keep him going”. According to the German military, the euphoric side effects of oxycodone were greater than those of morphine and the other semisynthetic opioids available at the time and it produced very powerful analgesia. Hence, they viewed it as a useful accompaniment to surgery on the battlefield. The use of Eukodal/oxycodone continued following the war. As we have seen from William Burrough’s comments above, it was the major opioid drug he consumed when he was living in Tangier and Paris prior to taking the apomorphine cure.
Generally speaking, up until the 1980s, opioids were used in medicine as part of anesthesiology during surgery and for treating acute pain resulting from injury. The only time when they were widely used for treating chronic pain was with cancer patients. Opioids were known to be highly addictive and therefore dangerous to use over extended periods of time unless there was no alternative. Used appropriately however they were fantastically effective drugs. These attitudes changed in the 1980s for a variety of reasons. Historians of the story of opioid drugs like to point to a short letter published by Porter and Jick in the New England Journal of Medicine (NEJM) in 1980 claiming that opioid addiction was very rare when opioids were used to treat people who were genuinely experiencing pain; it was only when they were used recreationally that they tended to become addictive. As a young faculty member in the Pharmacology Department of the University of Chicago in the 1980s I heard this assertation repeated by many of my medical colleagues. The general consensus was that pain was being undertreated and that the “American People” had a right to be free of pain. Why live with chronic pain caused by numerous diseases if you could be prescribed opioids and were unlikely to become addicted to them? The medical profession went so far as to designate pain as the “fifth vital sign”, a basic indicator as to the state of your health like blood pressure and heart rate. For reasons such as these there was a considerable upswing in the number of prescriptions for opioids for the treatment of all kinds of pain associated conditions. As it turns out, opioids aren’t particularly effective at treating conditions like chronic neuropathic pain. On the other hand, in contrast to letters in the NEJM and other claims, they are certainly addictive under all circumstances. What is more, pharmaceutical companies saw a wonderful opportunity for marketing drugs for treating “the fifth vital sign” in patients who had chronic pain conditions. One of these was Purdue Pharmaceuticals who had several advantages in the battle for the opioid market. Some members of the company had a great deal of experience in the advertising profession and had played a seminal role in developing modern methods of marketing drugs to doctors. Moreover, the company had already produced a version of morphine formulated so it was very long lasting and only had to be taken a couple of times a day, certainly a convenience if you were faced with taking the drug over an extended period of time. The patent position with morphine wasn’t very strong and so the company decided to apply all its marketing skills to an equivalent opioid drug; they chose oxycodone/Eukadol which, as we have seen, had been widely used for some time, and marketed a long-lasting version of the drug named which they named Oxycontin-soon to become known as “Oxy”. The inappropriate marketing of Oxycontin has been discussed in many other places. Suffice it to say that Oxy became a phenomenally successful and widely overprescribed drug. Unfortunately, a large number of people who took it became addicts. This was the first phase in what became known as the Opioid Epidemic.
As time went on and the extent of the Oxy addiction problem started to become more apparent, the number of opioid prescriptions began to fall once more and the price of Oxy on the black market began to rise until it was actually higher than that of heroin. Of course, most of the people who had become addicted to Oxy had encountered the drug through their doctors, not through drug dealers or other individuals outside the law. But once addicted they would unfortunately be only too willing to try anything to satisfy their craving. Moreover, many of these people had stored up excess medication that was unused because it had been overprescribed in the first place. It was therefore all too easy to sell Oxy and use the money to buy heroin and many of the Oxy addicts became street heroin addicts. This, of course, was accompanied by problems which Oxy addicts would not have previously encountered. These were not only problems with needles and infections associated with by drugs illegally. Some of the problems were also associated with the product that they were sold on the streets which could be of low quality or perhaps in quantities which were not what they were supposed to be. It is common for people using street heroin or other opioids to inadvertently overdose and die from drug induced respiratory depression. And it was for this reason that the epidemic was about to take an even more serious turn.
Prior to the start of the Second World War the pharmaceutical companies in Germany had been combined by the Nazi government to form a giant cartel named IG Farben (IGF or Interessen Gemeinschaft Farbenindustrie- “The combined interests of the dye making companies”). IGF subsequently played a key role as part of the chemical industry that supported Hitler’s war efforts. In 1937 a chemist named Otto Eisleb was working on a project for IGF attempting to make new antispasmodic drugs for treating bowel diseases. The idea here was to make new drugs that could block the effects of the neurotransmitter acetylcholine which normally initiates spasms in the colon and other areas of the gastrointestinal tract. Eisleb made several new molecules and passed them on to his pharmacology colleague Otto Schaumann for testing. One molecule in particular was an effective, but not very potent, antispasmodic. However, this molecule also produced another effect. It made mice stick their tails up in the air. This kind of behavior is quite obvious and is known as the Straub tail phenomenon. It is typical of morphine like analgesic drugs. Schaumann went on to test the molecule as an analgesic. Lo and behold it turned out to be extremely effective. The new molecule was named pethidine (it is also named meperidine or Demerol). Now, consider, that up until this time all new morphine derivatives had been obtained by performing chemical reactions on morphine, codeine or thebaine which are relatively difficult to obtain; they must be sourced from poppies. All the new opioid drugs that had been made were semi-synthetic derivatives of naturally occurring drugs. Pethidine was something completely new. It was synthesized “from scratch”, from basic materials that had never been anywhere near a poppy, and it wasn’t intended to be an analgesic. This property was discovered inadvertently. To begin with, it wasn’t clear why it should work like morphine when from the chemical point of view, it didn’t resemble it at all, having a much simpler chemical structure than traditional opioid drugs. But the thing is if you look at it properly it really does look like morphine (Figure 9). The structure of pethidine is called a phenylpiperidine and is clearly represented in the backbone of the morphine molecule, like dopamine being represented in the structure of apomorphine. It just turned out that although Nature had made morphine for some purpose that we still don’t understand (plants don’t have a nervous system and feel pain in any way we suppose), for its analgesic effects to be manifest in humans, only a small portion of the molecule was necessary, and this was the portion represented by pethidine. It is relatively easy for a chemist to make molecules like pethidine. You don’t need morphine or thebaine as a starting point; you just need some very basic materials and to perform some fairly rudimentary chemistry and you have a molecule that rivals what Nature has provided to us.

What’s more it is easy to make thousands of new molecules that look like pethidine and see what happens just by adding a new atom here or there. Perhaps they would not be addictive, depress respiration or have any of the poisonous side effects of morphine? As usual in the opioid field, that was the initial hope. Unfortunately, they are addictive -very addictive as a matter of fact. Pethidine became one of the most widely used drugs in the world and pethidine’s children and grandchildren have found a several important therapeutic niches in medicine. The development of such molecules came in stages. First of all, IGF started to play around with pethidine and during the war also produced methadone, another completely synthetic morphine like drug that has found utility in opioid reduction therapies for treating addicts. Addicts are given methadone under supervision as a substitute for a drug like heroin and gradually weaned off it. The structure of methadone is similar to that of pethidine and, again, is relatively easy to synthesize from scratch. After the war, in the 1950s, a highly innovative drug company in Belgium known as Janssen Pharmaceutica began a program to develop pethidine like analgesics. The head of the company, Paul Janssen, had the idea that if he could produce a super-potent analgesic drug, it might have fewer non-analgesic side effects. He would be disappointed as far as that was concerned-but the super-potent drugs were relatively fast in appearing. Like others at the time Janssen started to riff off the structures of pethidine and methadone and in 1956 discovered the methadone analogue dextromoramide (Palfium).Turning his attention to the pethidine structure in 1960 he synthesized fentanyl, a molecule that was approximately 100 times more potent that morphine.* Fentanyl soon became the analgesic of choice for many anesthesiologists owing to its rapid onset, short duration of action, high potency and limited cardiovascular side effect profile. Over the next twenty years hundreds of fentanyl analogues followed, some of which proved useful to anesthesiologists; many of them were extraordinarily potent. Sufentanil, carfentanil, alfentanil and ohmefentanyl-some of these drugs are up to 30,000 times as potent as morphine. That is not a typo-30,000 times!
(*It is also interesting to note that as well as using pethidine as a basis for creating fentanyl, Janssen also used it to create a completely different type of drug -haloperidol, one of the first and most effective antipsychotic drugs for the treatment of schizophrenia.)
Although fentanyl and its analogues have found a place in medicine and anesthesiology, they have also had some extremely unfortunate consequences. All these drugs act in the same way as more traditional opioids such as morphine, heroin and Oxy. Their mechanism is to activate a cellular receptor protein called the m-opioid receptor which is expressed by certain nerve cells specifically located throughout the neuraxis. Consequently, they are perfectly acceptable alternatives for an addict who wants to get high. Looked at another way, it would be much easier for a drug dealer to synthesize and distribute fentanyl than traditional morphine-like opioids. This idea took a little time to catch on, but as early as the 1970s fentanyl abuse started to appear as an alternative to heroin. Now these drugs have become central to the increased number of deaths seen during the opioid epidemic. As we have discussed, fentanyl and its analogues are not only fairly easy to make but are hundreds or thousands of times more potent as morphine or heroin. Hence on a weight-by-weight basis you only need one percent or less fentanyl to get high in comparison with drugs like morphine or even heroin. Fentanyl and its analogues have become widely available as street drugs, entering the US mostly from illegal laboratories in China and other countries. Clearly a gram of fentanyl will go a long way and is not difficult to smuggle into the US either live or by mail, frequently as the result of contacts on the Internet. At some point drug pushers realized that instead of selling heroin they could just add a little fentanyl to some innocuous white powder and sell that instead as “heroin”. Or if not fentanyl, how about sufentanil, carfentanil……..? Imagine the consequences of doing this. If a thumbnail quantity of heroin might be enough to kill you, then it would only take a tiny amount of fentanyl to do the same thing. A single small crystal or even less, something that you cannot even see, of one of the super-potent fentanyl derivatives could easily kill you. Street pushers are not that careful about weighing out how much drug needs to go into each sample that they sell, particularly when it comes to tiny amounts. The result is that many of the samples of street fentanyl laced drugs have easily enough opioid in them to be extremely toxic and many people have died from inadvertently overdosing. Hence this current phase of the opioid epidemic is the most dangerous of all. Nevertheless, the story of the chemical transformation of morphine had one more twist.
Antagonizing opioids
When we look at the structure of morphine, we know that it contains a nitrogen atom, and this is what is responsible for its basic character making it an alkaloid. Normally, in morphine and codeine, this nitrogen atom has a -CH3 methyl group attached to it. What happens if we use chemistry to play around with this structural feature? Taking it off and just leaving a hydrogen atom (normorphine) drastically reduces the potency of both morphine and codeine, so it appears to be necessary for their potent opioid effects. But what happens if you put something else on the nitrogen instead? In 1915 Julius Pohl at the University of Breslau made the fascinating observation that if you changed the methyl group on codeine for another organic group called allyl the new substance had some very strange properties. It didn’t work nearly as well as codeine or morphine as an analgesic. Instead, it was very good at reversing the effects of morphine. In other words, if you gave N-allyl-norcodeine it would stop morphine or codeine from being an analgesic, it would stop them producing constipation and, very importantly, it would reverse the suppression of breathing produced by these drugs. It took a long time before people figured out what was going on. Indeed, it was almost 30 years later the equivalent morphine-based molecule N-allyl-normorphine, known as nalorphine, was produced, and was found to produce similarly striking effects. These substances were the first opioids with antagonist activity. As we have discussed, opioids normally produce their effects by activating a protein called the m-opioid receptor. The receptor is like a lock and the opioid is like a key. Once the key is inserted into the lock it can turn it on and elicit important biological events in nerve cells. But what would happen if you made a molecule that fitted tightly into the lock but didn’t turn it on? The entire system would now be blocked up and nonfunctional. Opioids like morphine could now no longer work because their access would be blocked. Such compounds, of which N-allylnorcodeine and N-allylnormorphine were the first exemplars are what are known as opioid antagonists. As time went on even more selective and more potent antagonists were synthesized, particularly starting, interestingly enough, with molecules like hydrocodone and oxycodone rather than morphine, producing naloxone (N-allylnoroxycodone) and naltrexone (N-cyclopropylnoroxycodone). These are very potent and specific antagonists of the effects of morphine and of heroin or fentanyl or any other opioid. They are a godsend for anybody who is dying from an opioid overdose. Administration of naloxone or naltrexone rapidly reverses opioid induced respiratory depression and breathing is restored. These days, in the depths of the opioid epidemic, one frequently hears that first responders carry supplies of opioid antagonists so that they can be injected into victims and save their lives. And so, it can be seen two of morphine’s descendants, Oxy and its derivative naloxone, had very different fates in terms of their impact on humanity. It is interesting that, because of their physical properties, drugs like naloxone and naltrexone only work if given intravenously and not by mouth, something that would have been of great interest to Sir Robert Boyle. It is a curious fact that although the use of organic chemistry has not succeeded in ridding opioids of their problematic effects, they have generated drugs like opioid antagonists and apomorphine that can be used to treat them.
Is the current opioid epidemic the inevitable result of the intrinsic properties of these drugs? That is clearly not the case. We have known about their dangers as well as their benefits for thousands of years. Rather, the opioid epidemic has been the result of inappropriate human behavior. Indeed, it’s just as well to remember that this isn’t the first opioid epidemic. In the nineteenth century there was a similar problem in China resulting from the fact that the British insisted on aggressively trading the opium they grew in India to pay off trade debts they had with the Chinese. Opium became extremely abundant in China and, just as in the 21st century, its widespread availability fueled a nationwide opioid epidemic. We seem to have conveniently forgotten about it. How could we be so ignorant and naïve to allow widespread prescription of opioid drugs when history makes it perfectly clear what would happen? Or was it just hubris; the fact that doctors and scientists said to themselves that they know so much more about medicine in the 21st century that such a thing wouldn’t happen. In the US aggressive marketing of opioid drugs was the most important factor that led to the current epidemic. It’s not that different from the behavior of the British in China in the 19th century. The drug companies that were responsible for this were provided with an opening due to the almost willful misreading by the medical community of an 800-word letter in the New England Journal of Medicine, the implications of which go beyond the current text and suggest systemic problems in the way scientific publications are read and represented to both doctors and the public. There are clearly lessons here for the future.
Robert Boyle’s aim of separating the beneficial and poisonous effects of morphine has still not been achieved. Nevertheless, a great deal has been learned about the chemistry and pharmacology of opioid drugs. Humans have used the tools of chemistry to alter what was initially a gift from Nature and change it in countless ways. A small change in structure can make a huge difference to the properties of the resulting molecule. Potent or weak, agonist or antagonist, opioid agonist or dopamine agonist. The story of morphine illustrates the potential consequences when humans begin to alter ancient natural products using their modern chemical skills. And, sure enough, the story of morphine has been repeated numerous times.